Introduction
Input Data Format
Uploading and Processing
Obtaining an account
Upload Form
Data uploading
Processing Datasets
Project Management
Browsing Pathways
Promoter analysis
Analyzing Dataset
Changed Pathways
GO Enrichment
Functional classification
Searching
Adding New Platforms
Update Pathways and Genes
Contacts
|
Analyzing Dataset
Plant MetGenMAP provides three tools to further explore and analyze the uploaded datasets:
1) Identify significantly changed pathways; 2) Identify enriched Gene Ontology (GO) terms;
3) Functional classification of a list of genes.
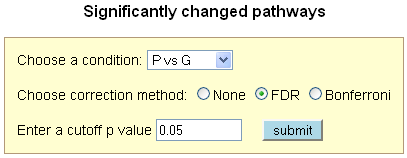
Identify Significantly Changed Pathways
Since in each condition, the statistical test (hypergeometric test) to check the significance
of pathway changes is performed on a set of pathways simultaneously, the raw p values need to
be corrected for multiple testing. Plant MetGenMAP provides the most commonly used multiple
test correction method - False Discovery Rate (FDR), as well as Bonferroni correction method.
Based on the user specified parameters, a list of significantly changed pathways is returned.
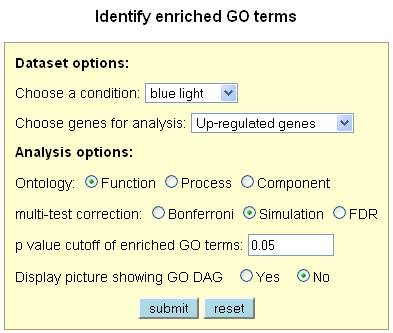
Identify Enriched GO Terms
This tool allows the user to identify over-represented (enriched) GO terms from a list of up-
and/or down-regulated genes under a specific condition. The tool was implemented based on the
GO::TermFinder perl module
(
Boyle et al., 2004) which uses the hypergeometric distribution to calculate the significance
of GO term enrichment. The tool provides three types of multiple test correction methods that
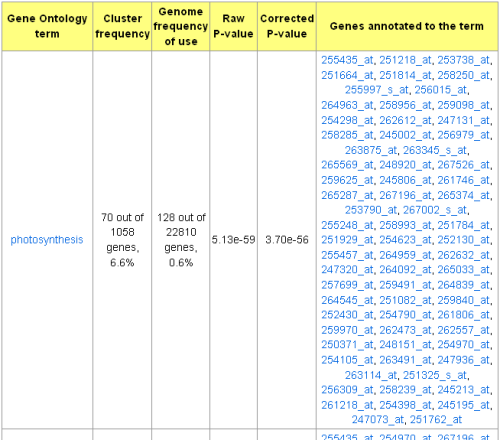
come with the GO::TermFinder module: FDR, Simulation, and Bonferroni. Significantly enriched
GO terms, genes annotated to each corresponding GO term, and the GO DAG picture (optional) are
included in the output page.
Note: This program may take several minutes if the number of input
genes is large.
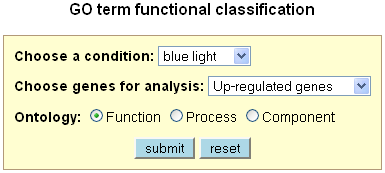
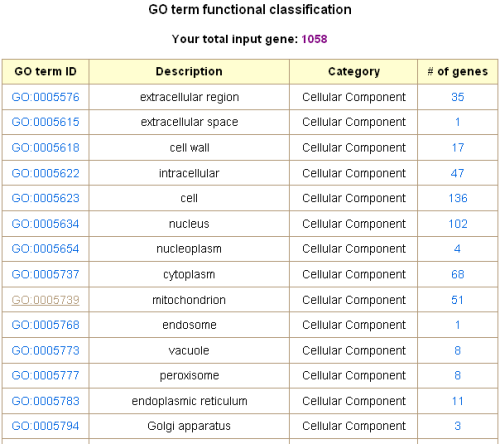
Gene functional classification
This tool classifies a list of interesting genes into different functional categories based on
their GO annotations. The functional catalogue is based on the Plant GO Slim, a cut-down version of the plant GO ontologies containing a
subset of the terms in the whole GO. These GO slims give a broad overview of the ontology
content without the detail of the specific fine grained terms.
From a list of up- and/or down-regulated genes under a specific condition, the tool outputs
the number of genes in each GO slim category with each number linked to the full list of
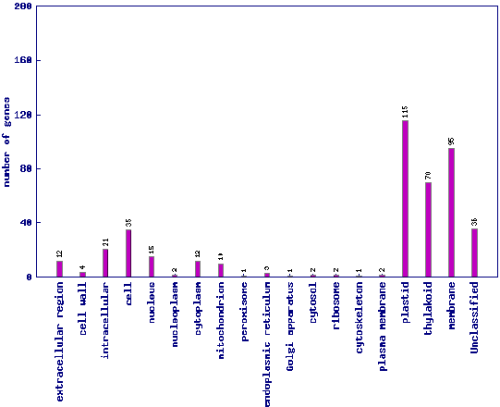
genes belonging to the corresponding category. A clickable bar graph is also provided. It
is worth noting that one gene could be assigned to several different GO slims, so the sum
of genes assigned to each category could be more than the number of total input genes.
|






