SVFilter
Introduction
Genomic structural variations (SVs), including large deletions, insertions, inversions, duplications and translocations, constitute an
important source of genetic diversity. Recent advances in next-generation sequencing (NGS) technologies and computational algorithms
have enabled the genome-wide mapping of SVs at a fine resolution. However, false discovery rate in the current SV discovery programs
remains high. We have developed the following five filters that can be used to efficiently identify false SVs.
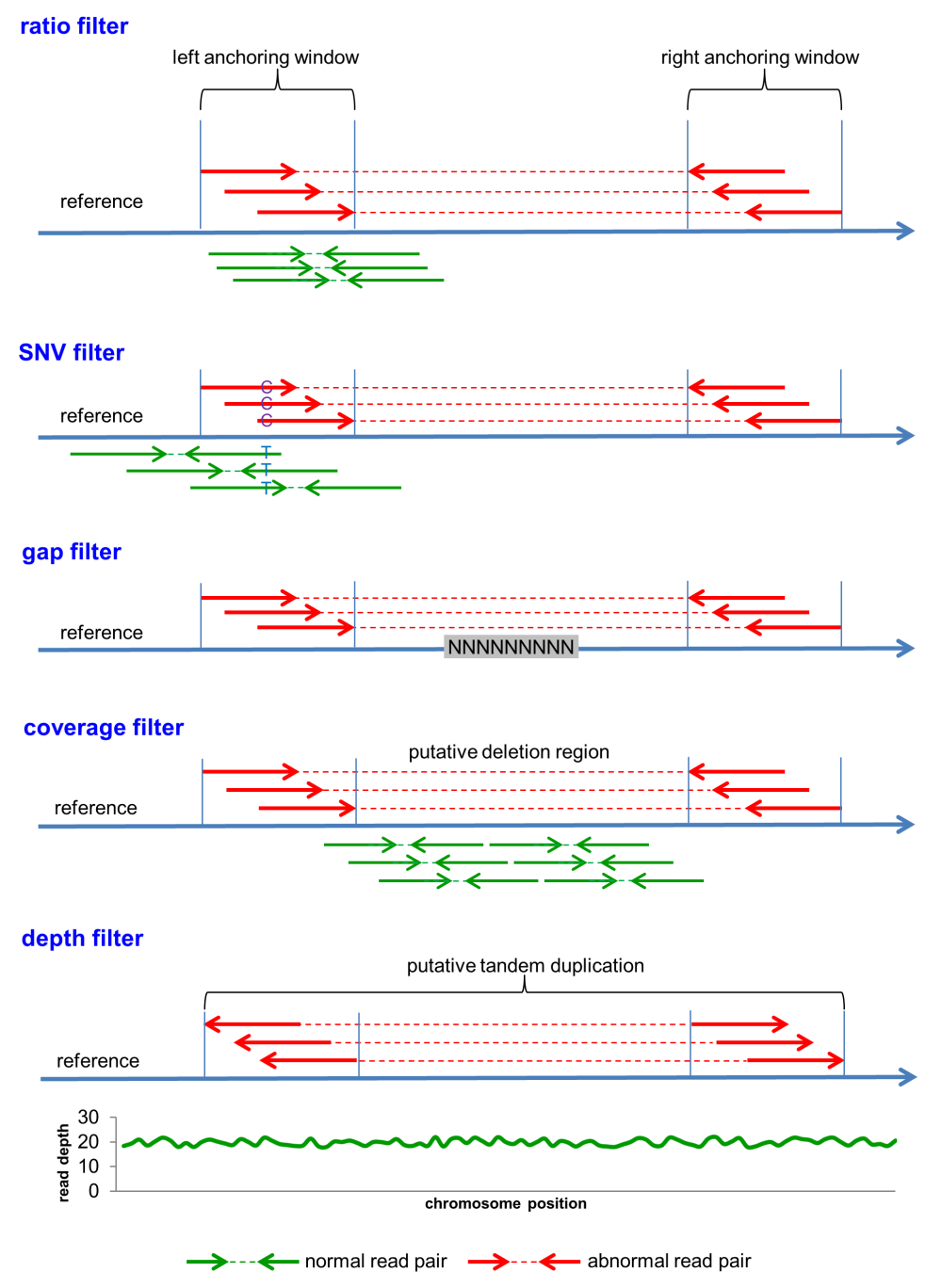
- Ratio filter - filter based on the ratio of normal and abnormal reads. Within an anchoring window
where abnormal reads cluster and form a SV, if substantial normal reads, which share the same orientation as the abnormal reads, are also
present, then this SV is discarded.
- SNV filter - filter based on the SNVs between normal and abnormal reads. Within an anchoring window,
if SNV(s) can be detected between normal and abnormal reads, then this SV is removed.
- Gap filter - filter based on gaps in the identified SVs. If an SV spans a genomic region
that contains gap(s), then this SV is discarded. Such SV spans across at least two contigs or even two scaffolds.
- Read coverage filter - filter based on the read coverage of the potential SV by normal reads. This
filter can only be applied to deletion events. If a substantial fraction of the deleted region is covered by normal reads, then this
candidate deletion is removed.
- Sequencing depth filter - filter based on the sequencing depth of the potential SV region. This filter
can be applied to tandem duplication events. If the average sequencing depth over the duplicated segment is not significantly higher than
the genome-wide average, then the predicted tandem duplication is rejected.
Please check this figure for further explanation of these filters.
System requirement and dependencies
- Linux (required) - Mac OS X is not supported
- samtools
Installation
Download the SVFilter and unzip the downloaded file.
| $ tar -xzvf SVFilter-1.0.tar.gz
|
This will generate a directory named "SVFilter-1.0". The directory contains three subdirectories:
- bin directory: includes all executables.
- test_files directory: includes all necessary input files to test the SV filter programs
- src directory: includes C++ source codes.
The executables under the "bin" directory were pre-compiled on a 64-bit Linux machine. For a 32-bit Linux machine, user needs to compile
the C++ source codes and then moves the executables to the "bin" directory. This can be done by running "install.sh" shell script
(sh install.sh) provided in the package.
Next, add the "bin" directory to the environmental variable PATH.
SV file format
Each of the SV filters requires an input file in tab-delimited text format which contains a list of SVs (deletion, insertion, inversion, duplication, etc.).
Here is an example of the file. Each SV contains 11 fields. Here is the explanation of each field:
| field No. | field name | example value | notes |
|---|
| 1 | chromosome/scaffold ID | chr1 | These four fields define the left anchoring window
of the SV: where it locates (chromosome/scaffold and start and end coordinates), and orientation of the abnormal reads mapped within the window:
R for reverse, F for forward |
| 2 | start position | 18099267 |
| 3 | end position | 18099607 |
| 4 | read strand | R |
| 5 | chromosome/scaffold ID | chr1 | These four fields define the right anchoring window of the SV |
| 6 | start position | 18100733 |
| 7 | end position | 18101053 |
| 8 | read strand | F |
| 9 | number of abnormal pairs | 4 | |
| 10 | abnormal read IDs | (FC42CA5AAXX:5:51:1514:1044#0, FC42CA5AAXX:5:95:188:1715#0, FC42CA5AAXX:5:23:1419:1684#0,
FC42CA5AAXX:5:78:1132:1599#0) | This field lists the IDs of abnormal read pairs, one for each pair. The IDs must be enclosed within a parenthesis and
separated by commas |
| 11 | SV type | DELETION | The type of the SV including DELETION, INSERTION, INVERSION and LARGE_DUPLI |
Run SVFilter
The five filters implemented in SVFilter are run separately. Each filter generates two output files, one containing the list of SVs that pass the filter (kept)
and one containing the list of SVs that are discarded by the filter. The file containing the kept SVs is in the format described above and can be
used as the input for other filters. Click here for detailed description of the files containing discarded SVs.
Ratio filter - run the program:
| $ ratiofilter test_SV normPair.sam 0.2 75
|
Explanation of the parameters
| parameter | example value | description |
|---|
| 1 | test_SV | SV input file containing a list of SVs in a format described above. |
| 2 | normPair.sam | File containing the alignments of normal paired-end reads in SAM format. |
| 3 | 0.2 | Cutoff ratio between the numbers of normal reads and abnormal reads within an anchoring window. The normal
reads must have the same orientations as abnormal reads. The SV input file specifies number of abnormal read pairs supporting each SV. Within one (or both)
of the two anchoring windows, if the ratio between the number of normal reads and abnormal reads with same orientations exceeds the cutoff value, then the
SV is considered as a false positive and discarded. |
| 4 | 75 | Read length (bp) |
SNV filter - run the program:
| $ SNVfilter test_sv genome.fa abnorm-pair.sam normPair.sam 2
|
Explanation of the parameters
| parameter | example value | description |
|---|
| 1 | test_SV | SV input file containing a list of SVs in a format described above. |
| 2 | genome.fa | Sequences of the reference genome in fasta format. |
| 3 | abnormPair.sam | File containing the alignments of abnormal paired-end reads in SAM format. |
| 4 | normPair.sam | File containing the alignments of normal paired-end reads in SAM format. |
| 5 | 2 | Minimum read depth to detect SNVs between abnormal and normal reads. If there is at least one SNV
detected between abnormal and normal reads in one (or both) anchoring window, the SV is considered as a false positive and discarded. |
gap filter - run the program:
| $ gapfilter test_sv genome.fa 1 0.1
|
Explanation of the parameters
| parameter | example value | description |
|---|
| 1 | test_SV | SV input file containing a list of SVs in a format described above. |
| 2 | genome.fa | Sequences of the reference genome in fasta format. |
| 3 | 1 | Cutoff of gap size (number of 'N') within the SV. |
| 4 | 0.1 | Minimum fraction of the gap in the SV region (ratio between gap size and SV size).
Assuming the size
of an SV is 2500 bp and within the SV region 300 bp are 'N's (gaps), then this SV will be discarded since 300 > 1 and 300 / 2500 > 0.1. If parameter 3 is
set to 1 and parameter 4 is set to a negative value, then the SV will be discarded as long as there is a gap within the SV region. |
read coverage filter - run the program (this filter is only applicable to deletion events):
| $ coveragefilter test_sv genome.fa test.pileup 6 0.05 2
|
Explanation of the parameters
| parameter | example value | description |
|---|
| 1 | test_SV | SV input file containing a list of SVs in a format described above. |
| 2 | genome.fa | Sequences of the reference genome in fasta format. |
| 3 | test.pileup | Pileup file generated only from normal reads. The file is used to check whether the putative
deleted region contains normal mapped reads. The file is generated from BAM file using the "samtools mpileup" utility. |
| 4 | 6 | Minimum number of base pairs in the deleted region that are mapped by normal reads. |
| 5 | 0.05 | Minimum fraction of the deletion region that are mapped by normal reads |
| 6 | 2 | Minimum depth of normal reads in the deleted region.
Assuming the length of a deleted region is 2500 bp
and within the deleted region 300 bp are covered by normal reads with at least 2X, then this SV will be discarded since 300 > 6 and 300 / 2500 > 0.05. |
sequencing depth filter - run the program:
| $ depthfilter test_sv chr-length test.pileup 1.5 1.5
|
Explanation of the parameters
| parameter | example value | description |
|---|
| 1 | test_SV | SV input file containing a list of SVs in a format described above. |
| 2 | chr-length | The file lists the length for each chromosome/scaffold. Each line consists of chromosome/scaffold ID
and its length, delimited by tab. |
| 3 | test.pileup | Pileup file generated only from normal reads. The file is used to calculate sequencing depths. The
file is generated from BAM file using the "samtools mpileup" utility. |
| 4 | 1.5 | Minimum ratio between the average of sequencing depth in the duplicated region and that over entire genome. |
| 5 | 1.5 | Minimum ratio between the median of sequencing depth in the duplicated region and that over the entire genome.
The filter calculates the average and median of sequencing depth of a duplication region, as well as the entire genome. If the ratio of the average depths between the
duplicated region and the entire genome < cutoff (1.5), or the ratio of the median depth < cutoff (1.5), then this duplication event is discarded. |
Frequent Asked Questions (FAQs)
- I obtained a set of SVs using breakpoint-based approaches such as Pindel. Can I use
SVFilter to identify potential false SVs?
Answer: Yes, SVFilter can be used. In this case, start and end positions of left and right anchoring windows will be collapsed to left and right breakpoints, respectively.
Since both anchoring windows are collapsed into single positions, ratio and SNV filters can not be used, while gap, coverage and depth filters can be applied to the SVs identified
with breakpoint-based approaches. In the SV input file, it's not necessary to specify read IDs. You can just put "(NA)" in the corresponding field. Here is an example:
| Chr1 | 884482 | 884482 | F | Chr1 | 886071 | 886071 | R | 6 | (NA) | DELETION |
- Do I have to provide read IDs in the SV input file?
Answer: Only SNV filter requires read IDs because it has to derive genotype information from the abnormal reads. Read IDs are not required for the other four filters
and you can just put "(NA)" in the read ID field for these four filters.
Download
Download SVFilter from the ftp server
Contact
For questions and suggestions, please contact us at feibioinfolab@gmail.com
|
|
{kind=link}